3. Constraints-based Flux analysis¶
3.1. Basic Mass Balance-constrained Analysis (FBA)¶
ETFBA package is capable of performing constraints-based flux analysis, incorporating constraints on enzyme protein allocation and/or reaction thermodynamics. Let’s start with the fundamental flux balance analysis (FBA). FBA aims to solve the following problem:

Here, \({\bf{S}}\) is the stoichiometric matrix, and \({\bf{v}}\) denotes the \({\bf{total}}\) \({\bf{fluxes}}\), i.e., the net flux through a reversible reaction is split into forward and backward (reverse) fluxes, both maintaining a non-negative value.
As a demonstration, we utilize the translated E. coli model iML1515 to conduct basic FBA.
[1]:
from etfba import Model
model_file = '../../models/e_coli/etfba_iML1515.bin'
model = Model.load(model_file)
print(model)
model iML1515 with 2712 reactions and 1877 metabolites
[2]:
objective = {'BIOMASS_Ec_iML1515_core_75p37M': 1}
flux_bound = (0, 1000)
spec_flux_bound = {'ATPM': (6.86, 1000)}
preset_flux = {'EX_glc__D_e_b': 10, 'FHL': 0}
res = model.optimize(
'fba',
objective=objective,
flux_bound=flux_bound,
spec_flux_bound=spec_flux_bound,
preset_flux=preset_flux
).solve(solver='gurobi')
The objective of our flux balance analysis can be set through the “objective” argument, which accepts a dictionary defining the expression of the objective function which is defined as the linear combination of fluxes. For example, {‘v1’: 2, ‘v2’: -1} defines the expression “2*v1 - v2”. “flux_bound” is used to set the bounds for all fluxes, while it is prioritized by “spec_flux_bound,” which can be used to set the bounds of specific fluxes. “preset_flux” is used to set fixed values of fluxes such as measured exchange rates or silenced reactions.
Note: 1. Fluxes are in the unit of mmol gCDW\(^{-1}\) \(h^{-1}\); 2. Since total fluxes are used as decision variables in the problem, a suffix of ‘_f’ or ‘_b’ is required to indicate forward or backward flux for reversible reactions in the above setting of arguments.
The objecitve at solution can be accessed by the opt_objective attributute of the optimization result:
[3]:
print('Optimal growth rate:', round(res.opt_objective, 3))
Optimal growth rate: 0.877
The net reaction fluxes derived from the solution can be accessed by the opt_fluxes attribute, which supports a save method for direct result saving.
[4]:
print('Top 10 reactions with the highest fluxes:')
for rxnid, flux in sorted(res.opt_fluxes.items(),
key=lambda item: item[1], reverse=True)[:10]:
print(f'{rxnid:>10} {flux:6.3f}')
Top 10 reactions with the highest fluxes:
HPYRRx 1000.000
FE2tex 1000.000
CRNt8pp 1000.000
CRNDt2rpp 1000.000
GLBRAN2 1000.000
GLUt4pp 1000.000
MN2tpp 1000.000
GLDBRAN2 1000.000
FUMt1pp 1000.000
R1PK 999.999
In naive FBA, some reactions may receive unseasonably high fluxes (as shown in the reactions above with the highest fluxes). This can be impractical in terms of resource economy during cell growth. parsimonious FBA can be used to address the problem by minimizing the overall fluxes while maintaining the optimality of the objective to some extent. Parsimonious FBA can be formulated as:
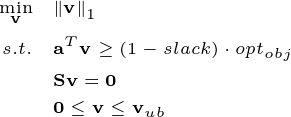
Here, a small constant \(slack\) is provided to relax the objective constraint in case it encounts difficiulties in finding feasible solutions.
Setting the “parsimonious” argument to True will prune the obtained fluxes.
[5]:
res = model.optimize(
'fba',
objective=objective,
flux_bound=flux_bound,
spec_flux_bound=spec_flux_bound,
preset_flux=preset_flux,
slack=0,
parsimonious=True
).solve(solver='gurobi')
print('Optimal growth rate:', round(res.opt_objective, 3))
INFO: estimating parsimonious fluxes
Optimal growth rate: 0.877
In estimating the parsimonious fluxes, basic FBA will be performed first, followed by minimizing overall fluxes to obtain parsimonious fluxes. Now we can have a more reasonable intracellular fluxes.
[6]:
print('Top 10 reactions with the highest fluxes:')
for rxnid, flux in sorted(res.opt_fluxes.items(),
key=lambda item: item[1], reverse=True)[:10]:
print(f'{rxnid:>10} {flux:6.3f}')
Top 10 reactions with the highest fluxes:
ATPS4rpp 70.432
EX_h2o_e 47.162
CYTBO3_4pp 44.256
NADH16pp 37.997
EX_co2_e 24.003
O2tpp 22.132
O2tex 22.132
GAPD 17.105
ENO 15.599
GLCptspp 10.000
To check the balance of a specific metabolite, one can use the statement method, which summarizes the overall production and consumption reaction fluxes to and from that metabolite.
[7]:
for key, rxn_flux in res.statement('nadph_c').items():
print(key)
for rxnid, flux in rxn_flux.items():
if abs(flux) > 0.001:
print(f'{rxnid:>10} {flux:6.3f}')
productions
ICDHyr 6.913
GND 2.355
G6PDH2r 2.355
MTHFD 0.867
consumptions
SHK3Dr 0.334
G5SD 0.194
KARA1 -0.767
KARA2 0.255
GLUTRR 0.003
AGPR -0.259
DXPRIi 0.002
UAPGR 0.024
DHDPRy 0.325
TRDR 0.217
G3PD2 -0.122
P5CR 0.194
ASAD -0.938
SULR 0.217
HSDy -0.612
GLUDy -7.499
DHFR 0.024
3OAR140 0.068
Note: 1. The “statement” method summarizes fluxes of the reactions in which a metabolite is involved. To obtain a metabolite-based mass balance, the stoichiometric coefficient in corresponding reactions needs to be considered; 2. While fluxes are aggregated based on the production or consumption of a metabolite, the sign of the flux value depends on the directionality of the reaction as defined in the model.
3.2. Constrained by Both Enzyme Protein Allocation and Mass Balance (EFBA)¶
Metabolic reactions rely on enzymes to proceed. Higher fluxes usually require more enzyme amounts to catalyze the chemical process. Therefore, it is essential to incorporate enzyme protein constraints to optimize flux distributions under resource-limited conditions, which are common in realistic scenarios. EFBA can be formulated as:
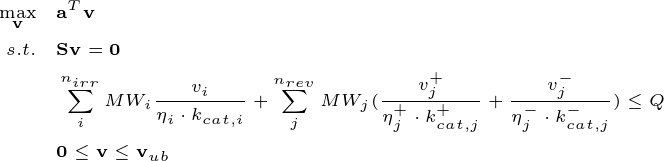
Here, \(n_{irr}\) and \(n_{rev}\) are the number of irreversible and reversible reactions, respectively. The superscipts “+” and “-” denote the forward and reverse reactions, respectively. \(\eta\) represents the enzyme efficiency affected by substrate saturation and thermodynamic feasibility. The product of \(\eta\) and \(k_{cat}\) forms the apparent enzyme catalytic constant, \(k_{app}\). \(Q\) denotes the total available protein resource fraction for metabolic enzymes in biomass (g gCDW\(^{-1}\)).
Let’s continue with the E. coli model with the enzyme protein allocation constraint added:
[8]:
import pandas as pd
kcat_file = '../../models/e_coli/kcats.xlsx'
mw_file = '../../models/e_coli/mws.xlsx'
kcats = pd.read_excel(kcat_file, header=None, index_col=0).squeeze()
mws = pd.read_excel(mw_file, header=None, index_col=0).squeeze()
# set reactions with available kcats and MWs
eff_rxns = []
for rxnid, rxn in model.reactions.items():
if rxn.rev:
if rxnid+'_f' in kcats.index and rxnid+'_b' in kcats.index and rxnid in mws.index:
rxn.forward_kcat = kcats[rxnid+'_f']
rxn.backward_kcat = kcats[rxnid+'_b']
rxn.molecular_weight = mws[rxnid]
eff_rxns.append(rxnid)
else:
if rxnid in kcats.index and rxnid in mws.index:
rxn.forward_kcat = kcats[rxnid]
eff_rxns.append(rxnid)
enz_ub = 0.15
preset_flux = {'FHL': 0}
res = model.optimize(
'efba',
objective=objective,
flux_bound=flux_bound,
spec_flux_bound=spec_flux_bound,
preset_flux=preset_flux,
inc_enz_cons=eff_rxns,
enz_prot_lb=enz_ub,
parsimonious=True # used to obtain parsimonious fluxes
).solve(solver='gurobi')
print('Optimal growth rate:', round(res.opt_objective, 3))
INFO: estimating parsimonious fluxes
Optimal growth rate: 0.866
The “inc_enz_cons” argument specifies reactions subject to the enzyme protein constraint.
Note that the substrate uptake is not set during optimization; the model will obtain the metabolic capability completely depending on the protein resource of catalytic enzymes.
[9]:
print('Glucose uptake rate:', -round(res.opt_fluxes['EX_glc__D_e'], 3))
Glucose uptake rate: 12.887
The optimal protein fractions allocated for each enzyme subjected to the constraint can be accessed by the opt_enzyme_costs attribute of the optimization result, which also has a save method for direct saving.
[10]:
print('Top 10 reactions with the most abundant enzyme protein allocations:')
for rxnid, pro in sorted(res.opt_enzyme_costs.items(),
key=lambda item: item[1], reverse=True)[:10]:
print(f'{rxnid:>10} {round(pro, 5):>10}')
Top 10 enyzme with the most abundant protein allocations:
ENO 0.01082
GAPD 0.00746
FBA 0.00623
PGK 0.00596
KARA2 0.00503
KARA1 0.00494
METS 0.00479
GHMT2r 0.00361
PGM 0.00357
GND 0.0027
3.3. Constrained by Both Thermodynamics and Mass Balance (TFBA)¶
To enforce the second law of thermodynamics, constraints on the reaction Gibbs energy change can be added to the model, ensuring reactions proceed in the thermodynamically favorable direction. The TFBA problem can be formulated as:
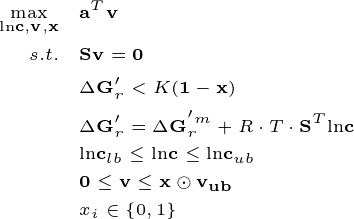
Here, metabolite concentrations \({\bf{c}}\) (log transformed) are introduced as new varibles, as well as the binary \({\bf{x}}\) (1 indicates nonzero flux, and 0 otherwise), making the problem a mixed-integer linear programming (MILP). \(K\) is big enough constant ensuring the thermodynamic constraints are applied only to nonzero fluxes.
We continue to use the E. coli model for demonstration.
[11]:
dgpm_file = '../../models/e_coli/dgpms.xlsx'
dgpms = pd.read_excel(dgpm_file, header=None, index_col=0).squeeze()
preset_flux = {'EX_glc__D_e_b': 10, 'FHL': 0}
conc_bound = (0.0001, 100)
spec_conc_bound = {
'o2_c': (0.0001, 0.0082), # should be less than in the media
'co2_c': (0.1, 100) # should be greater than in the media
}
preset_conc = {
'glc__D_p': 20,
'pi_p': 56,
'so4_p': 3,
'nh4_p': 19,
'na1_p': 160,
'k_p': 22,
'fe2_p': 62
}
# set reactions with available ΔG'm
ex_rxns = []
for rxnid, rxn in model.reactions.items():
if rxnid in dgpms.index:
rxn.standard_gibbs_energy = dgpms[rxnid]
else:
ex_rxns.append(rxnid)
res = model.optimize(
'tfba',
objective=objective,
flux_bound=flux_bound,
conc_bound=conc_bound,
spec_flux_bound=spec_flux_bound,
spec_conc_bound=spec_conc_bound,
preset_flux=preset_flux,
preset_conc=preset_conc,
ex_thermo_cons=ex_rxns,
parsimonious=True # used to obtain parsimonious fluxes
).solve(solver='gurobi')
print('Optimal growth rate:', round(res.opt_objective, 3))
INFO: estimating parsimonious fluxes
Optimal growth rate: 0.877
Obtaining the standard reaction Gibbs energy is a prerequisite to perform TFBA. The batch estimation of \(\Delta G'^0\) can be achieved with equilibrator-api which applies a component contribution method with improved accucary compared to canonical methods.
Note: Metabolite concentrations are provided in the unit of mM, thus the standard reaction Gibbs energ is actually \(\Delta G'^m\) which can be transformed from \(\Delta G'^0\) (see the calculation of \(\Delta G'^m\) in the Building a Model section).
The estimated reaction Gibbs energy, reaction directionality, and metabolite concentrations at the solution can be accessed with opt_gibbs_energy, opt_directions, and opt_concentrations attributes of the optimization result, which also have the save method.
[12]:
count = 0
for rxnid, dgp in res.opt_gibbs_energy.items():
if count <= 15:
rxn_dir = res.opt_directions[rxnid]
if rxn_dir != 'zero flux':
print(f'{rxnid:>10} {round(dgp,3):>10} {rxn_dir:>10}')
count += 1
SHK3Dr -46.227 forward
DHORTS 0.001 reverse
OMPDC -25.614 forward
G5SD -45.45 forward
CS -80.096 forward
ICDHyr -0.001 forward
PPA -47.042 forward
APRAUR -65.126 forward
TRPAS2 0.001 reverse
DB4PS -157.053 forward
ALAR -0.041 forward
RBFK -21.895 forward
ALATA_L 24.236 reverse
PPM -13.807 reverse
ASPTA 11.768 reverse
RBFSb -93.656 forward
3.4. Constrained by Enzyme Protein Allocation, Thermodynamics and Mass Balance (ETFBA)¶
Integrating all the above constraints of enzyme protein allocation, thermodynamics, and mass balance yields ETFBA, which allows a more comprehensive understanding of cell metabolism at the genome scale. As an optimization problem, it is defined as:
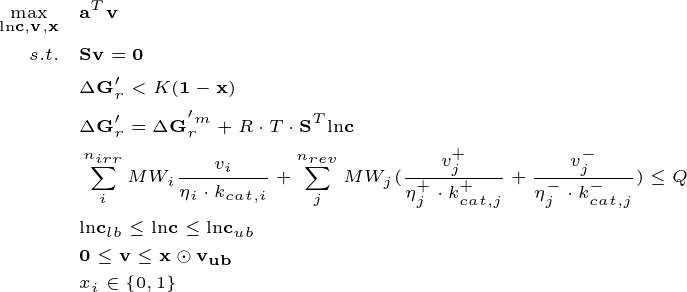
We are still using the E. coli model, but this time the subtrate uptake rate is not set.
[13]:
preset_flux = {'FHL': 0}
res = model.optimize(
'etfba',
objective=objective,
flux_bound=flux_bound,
conc_bound=conc_bound,
spec_flux_bound=spec_flux_bound,
spec_conc_bound=spec_conc_bound,
preset_flux=preset_flux,
preset_conc=preset_conc,
ex_thermo_cons=ex_rxns,
inc_enz_cons=eff_rxns,
enz_prot_lb=enz_ub,
parsimonious=True # used to obtain parsimonious fluxes
).solve(solver='gurobi')
print('Optimal growth rate:', round(res.opt_objective, 3))
INFO: estimating parsimonious fluxes
Optimal growth rate: 0.864
The result we obtained will have all the attributes mentioned above to access the fluxes, metabolite concentrations, enzyme protein costs, reaction Gibbs energies, reaction directionality, etc., as well as the statement method to inspect the balance of some metabolite at the solution.
[14]:
print('Top 10 reactions with the highest fluxes:')
for rxnid, flux in sorted(res.opt_fluxes.items(),
key=lambda item: item[1], reverse=True)[:10]:
print(f'{rxnid:>10} {flux:6.3f}')
Top 10 reactions with the highest fluxes:
ATPS4rpp 59.383
EX_h2o_e 42.636
CYTBO3_4pp 35.362
NADH16pp 35.075
GAPD 21.880
ENO 20.444
EX_co2_e 19.832
EX_h_e 18.897
O2tpp 17.990
O2tex 17.990